
Computational Pharmacology Lab

The Stockner Lab is an interdisciplinary research team that investigates membrane protein function by synergistically integrating computational and experimental approaches. We are passionate about achieving a comprehensive understanding for protein function. To reach this goal, we use approaches that collectively allow for obtaining insights, which none of the approaches could obtain on their own: We use MD simulation, advanced analyses methods and build on Machine Learning approaches assembled as a computational microscope to study membrane protein function with atom resolution. To these computational methods we add experimental approaches, most importantly classical pharmacology and electrophysiological measurements to obtain a comprehensive understanding of protein function at multiple levels. We carry out experiments in house, but also (quite frequently) employ collaborations with experimental groups.
Research Highlights
Please find below some of the recent research highlights!

Researcher of the Month
Ralph Gradisch earned with his work on substrate occlusion () by the serotonin transporter (SERT) the prestigous award, reseracher of the month.
Researcher of the Month, Oct. 2025
The “Researcher of the Month” jury has awarded this month's prize to Dr. Ralph Gradisch for his paper “Ligand coupling mechanism of the human serotonin transporter differentiates substrates from inhibitors” (https://doi.org/10.1038/s41467-023-44637-6). The multidisciplinary study was conducted as part of Dr. Ralph Gradisch's PhD studies at the Institute of Pharmacology in the group of Assoc. Prof. Dr. T. Stockner in collaboration with the groups of H. Sitte (Medical University of Vienna, Institute of Pharmacology), Marko Mihovilovic (Vienna University of Technology, Institute of Applied Synthetic Chemistry) and Randy D. Blakely (Florida Atlantic University, Department of Biomedical Science, Jupiter, FL, USA).

Free energy profile of the substrate-induced occlusion of the human serotonin transporter.
Alves da Silva L, Lazzarin E, Gradisch R, Clarke A, Stockner T.
J Neurochem. 2024 doi: 10.1111/jnc.16061.
https://onlinelibrary.wiley.com/doi/10.1111/jnc.16061
The serotonin transporter (SERT) is a member of the Solute Carrier 6 (SLC6) family and is responsible for maintaining the appropriate level of serotonin in the brain. Dysfunction of SERT has been linked to several neuropsychiatric disorders, including depression, anxiety and obsessive-compulsive disorder. Therefore, an in-depth understanding of the mechanism on an atomistic level, coupled with a quantification of transporter dynamics and the associated free energies is required. Here, we constructed Markov state models (MSMs) from extensive unbiased molecular dynamics simulations to quantify the free energy profile of serotonin (5HT) triggered SERT occlusion and explored the driving forces of the mechanism of occlusion. Our results reveal that SERT occludes via multiple intermediate conformations and show that the motion of occlusion is energetically downhill for the 5HT-bound transporter. Force distribution analyses show that the interactions of 5HT with the bundle domain are crucial. During occlusion, attractive forces steadily increase and pull on the bundle domain, which leads to SERT occlusion. Some interactions become repulsive upon full occlusion, suggesting that SERT creates pressure on 5HT to promote its movement towards the cytosol.
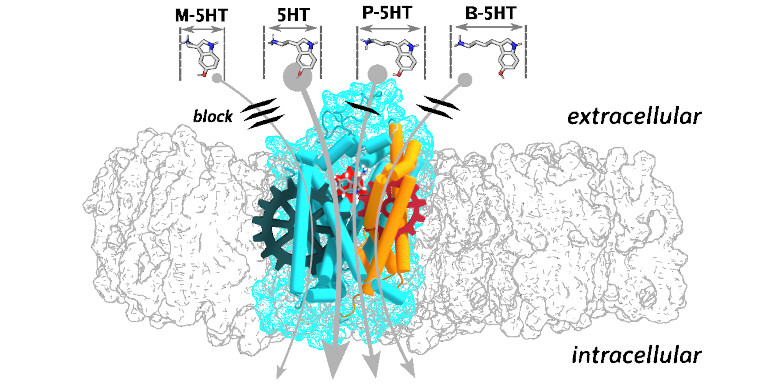
Ligand coupling mechanism of the human serotonin transporter differentiates substrates from inhibitors.
Gradisch R, Schlögl K, Lazzarin E, Niello M, Maier J, Mayer FP, Alves da Silva L, Skopec SMC, Blakely RD, Sitte HH, Mihovilovic MD, Stockner T. Nat Commun. 2024; 15(1):417. doi: 10.1038/s41467-023-44637-6.
https://www.nature.com/articles/s41467-023-44637-6
The presynaptic serotonin transporter (SERT) clears extracellular serotonin following vesicular release to ensure temporal and spatial regulation of serotonergic signalling and neurotransmitter homeostasis. Prescription drugs used to treat neurobehavioral disorders, including depression, anxiety, and obsessive-compulsive disorder, trap SERT by blocking the transport cycle. In contrast, illicit drugs of abuse like amphetamines reverse SERT directionality, causing serotonin efflux. Both processes result in increased extracellular serotonin levels. By combining molecular dynamics simulations with biochemical experiments and using a homologous series of serotonin analogues, we uncovered the coupling mechanism between the substrate and the transporter, which triggers the uptake of serotonin. Free energy analysis showed that only scaffold-bound substrates could initiate SERT occlusion through attractive long-range electrostatic interactions acting on the bundle domain. The associated spatial requirements define substrate and inhibitor properties, enabling additional possibilities for rational drug design approaches.

YASARA Model-Interactive Molecular Modeling from Two Dimensions to Virtual Realities.
Ozvoldik K, Stockner T, Krieger E. J Chem Inf Model. 2023;63(20):6177-6182. doi: 10.1021/acs.jcim.3c01136.
https://pubs.acs.org/doi/10.1021/acs.jcim.3c01136
The industry's transition from three-dimensional (3D) glasses to virtual reality (VR) headsets has left modelers stranded without hardware supply, since walking around and waving arms in a virtual world is a great experience but also very tiring when doing time-intensive modeling work. We present a novel software implementation that uses a VR headset while sitting at a desk in front of the normal screen, which is beamed into the virtual reality together with keyboard, mouse, and chair using the headset's cameras and an extra tracker attached to the seat-back. Compared to 3D glasses, this yields a comparably relaxing but much more immersive workplace and provides additional possibilities such as taking molecules into one's hands, standing up, and walking or teleporting through the models. This VR functionality has been combined with a molecular graphics engine based on Vulkan, a next-generation cross-platform application programming interface (API) for GPUs and the successor of the widely used Open Graphics Library (OpenGL). It is built into the YASARA Model program, which includes many features like small and large molecule builders, electron densities, partial surfaces, contact analysis, coordinate manipulation, and animations. Interactive tutorials are provided to guide modelers into VR and familiarize them with the molecular modeling features. YASARA Model is available for Linux, Windows, Android, and MacOS (the latter without VR) with an introductory video at www.YASARA.org/vr.
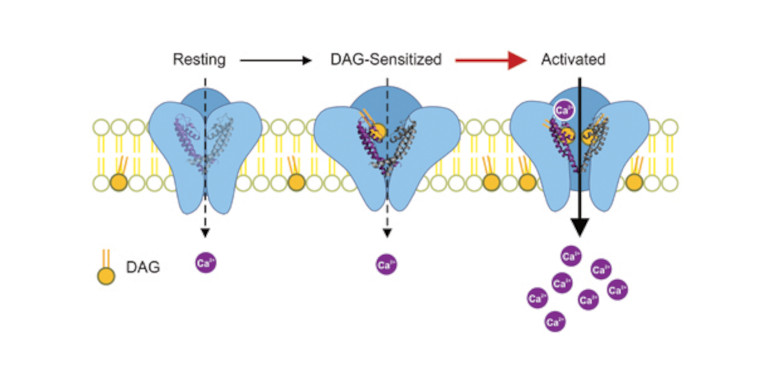
Diacylglycerols interact with the L2 lipidation site in TRPC3 to induce a sensitized channel state
Hazel Erkan-Candag, Amy Clarke, Oleksandra Tiapko, Mathias Af Gsell, Thomas Stockner, Klaus Groschner. EMBO Rep. 2022;23(7):e54276. doi: 10.15252/embr.202154276.
https://www.embopress.org/doi/full/10.15252/embr.202154276
Coordination of lipids within transient receptor potential canonical channels (TRPCs) is essential for their Ca2+ signaling function. Single particle cryo‐EM studies identified two lipid interaction sites, designated L1 and L2, which are proposed to accommodate diacylglycerols (DAGs). To explore the role of L1 and L2 in TRPC3 function, we combined structure‐guided mutagenesis and electrophysiological recording with molecular dynamics (MD) simulations. MD simulations indicate rapid DAG accumulation within both L1 and L2 upon its availability within the plasma membrane. Electrophysiological experiments using a photoswitchable DAG‐probe reveal potentiation of TRPC3 currents during repetitive activation by DAG. Importantly, initial DAG exposure generates a subsequently sensitized channel state that is associated with significantly faster activation kinetics. TRPC3 sensitization is specifically promoted by mutations within L2, with G652A exhibiting sensitization at very low levels of active DAG. We demonstrate the ability of TRPC3 to adopt a closed state conformation that features partial lipidation of L2 sites by DAG and enables fast activation of the channel by the phospholipase C‐DAG pathway.
Interaction of GAT1 with sodium ions: from efficient recruitment to stabilisation of substrate and conformation
Erika Lazzarin, Ralph Gradisch, Sophie M.C. Skopec, Leticia Alves da Silva, Dániel Szöllősi, Julian Maier, Sonja Sucic, Baruch I. Kanner, Harald H. Sitte, Thomas Stockner
doi: https://doi.org/10.1101/2023.10.10.561652
https://www.biorxiv.org/content/10.1101/2023.10.10.561652v1
The human GABA transporter (GAT1) is a membrane transporter that mediates the reuptake of the neurotransmitter GABA from the synaptic cleft into neurons and glial cells. Dysregulation of the transport cycle has been associated with epilepsy and neuropsychiatric disorders, highlighting the crucial role of the transporter in maintaining homeostasis of brain GABA levels. GAT1 is a secondary active transporter that couples the movement of substrate to the simultaneous transport of sodium and chloride ions along their electrochemical gradients. Using MD simulations, we identified a novel sodium recruiting site at the entrance to the outer vestibule, which attracts positively charged ions and increases the local sodium concentration, thereby indirectly increasing sodium affinity. Mutations of negatively charged residues at the recruiting site slowed the binding kinetics, while experimental data revealed a change in sodium dependency of GABA uptake and a reduction of sodium affinity. Simulation showed that sodium displays a higher affinity for the sodium binding site NA2, which plays a role in the stabilisation of the outward-open conformation. We directly show that the presence of a sodium ion bound to NA2 increases the stability of the closed inner gate and restrains motions of TM5. We find that sodium is only weakly bound to NA1 in the absence of GABA, while the presence of the substrate strengthens the interaction due to the completed ion coordinating shell, explaining cooperativity of between GABA and sodium.

Structural basis of organic cation transporter-3 inhibition
Basavraj Khanppnavar, Julian Maier, Freja Herborg, Ralph Gradisch, Erika Lazzarin, Dino Luethi, Jae-Won Yang, Chao Qi, Marion Holy, Kathrin Jäntsch, Oliver Kudlacek, Klaus Schicker, Thomas Werge, Ulrik Gether, Thomas Stockner, Volodymyr M Korkhov, Harald H Sitte
Nat Commun. 2022; 13(1):6714. doi: 10.1038/s41467-022-34284-8.
https://pubmed.ncbi.nlm.nih.gov/36344565/
Organic cation transporters (OCTs) facilitate the translocation of catecholamines, drugs and xenobiotics across the plasma membrane in various tissues throughout the human body. OCT3 plays a key role in low-affinity, high-capacity uptake of monoamines in most tissues including heart, brain and liver. Its deregulation plays a role in diseases. Despite its importance, the structural basis of OCT3 function and its inhibition has remained enigmatic. Here we describe the cryo-EM structure of human OCT3 at 3.2 Å resolution. Structures of OCT3 bound to two inhibitors, corticosterone and decynium-22, define the ligand binding pocket and reveal common features of major facilitator transporter inhibitors. In addition, we relate the functional characteristics of an extensive collection of previously uncharacterized human genetic variants to structural features, thereby providing a basis for understanding the impact of OCT3 polymorphisms.

Assoc.-Prof. Dr. Thomas Stockner
ORCID: 0000-0002-7071-8283
Tel.: +43 (0)1 40160-31215
E-Mail: thomas.stockner@meduniwien.ac.at