
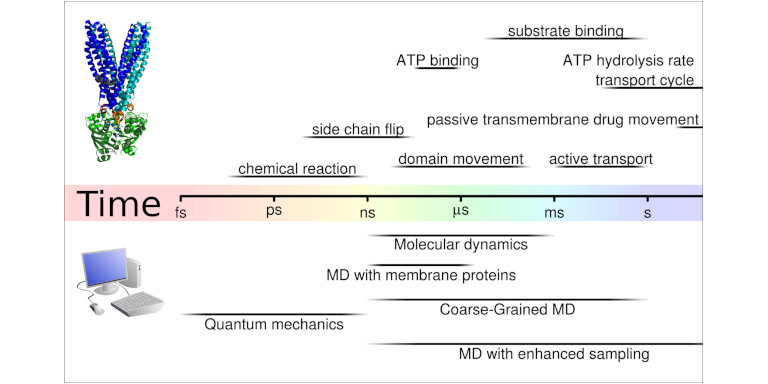
Methods
We implement and use different techniques of computational pharmacology, including molecular dynamics (MD) simulations, free energy and entropy calculations, but also apply structural bioinformatics methods and continue expanding the use of advanced Machine Learning approaches. These techniques are always used in close conjunction with experiments as we aim to discover new fundamental principles behind biological matter to explain essential biological phenomena from a quantitative, physicochemical and pharmacological perspective.
Computational Approaches
Enhanced sampling
Enhanced sampling to study slow processes in transporters
For MD simulations we use the very fast MD engine Gromacs (https://www.gromacs.org). Still, sufficient sampling remains for the near future a limiting factor for MD simulations of membrane proteins. All atom simulation can typically reach the low microseconds regime on computer hardware available at HPC clusters, while biological processes at membranes, such as substrate transport by secondary active transporters or channel opening of ligand gated ion channels, are in the low millisecond to second timescale. The long time scales of biological processes are dictated by energy barriers, often with large entropic components. Fortunately, not all is lost. Simulation theory has developed several approaches that allow for overcoming these time scale limitations. To overcome sampling limitations, long timescales and potential high energy barriers, we apply a number of enhanced sampling methods, most importantly well-tempered Metadynamics, Enforced Rotation, Steered Molecular Dynamics simulations, Conformational Flooding, Milestoning and Swarm simulations.
These methods add a bias in a controlled fashion to the underlying free energy surface to increase sampling in the directions of the reaction coordinate(s) of conformational transition. Well-tempered Metadynamics adds localized Gaussians to the free energy surface of the predefined reaction coordinates, thereby pushing the protein system along the predefined reaction path. Conformational Flooding destabilizes the starting conformation along all degrees of freedom, thereby inducing motions along the direction with the lowest energy barriers, which we can assume to coincide with the transition path. Similar to atomic force microscopy, Steered Molecular Dynamics simulations attach a virtual spring to the protein and exerts a pulling force to induce a conformational transition. Enforced Rotation is an approach that defines protein domains as rotational groups and defines a dynamically adjustable rotational axis. Protein domains are then rotated against each other, thereby e.g. allow transporters to switch conformation between inward-facing and outward-facing. Milestoning and Swarm simulations operate on the entropic part for enhancing the sampling by using a large number of short parallel simulations along the reaction coordinate, like pearls on a necklace. Each individual simulation performs only local sampling, but by using for example Markov Chain Analysis all these short simulation can be connect to collectively describe the entire transition path.
Free Energy of Conformational Changes
Protein motions and dynamics are governed by the underlying free energy. The underlying free energy surface of protein motions can be directly derived from well-tempered Metadynamics and from the accelerated weight histogram method (AWH), or complementary to these methods, by one of the earlier developed perturbation methods, the Potential of Mean Force (PMF) approach. We are also using Markov State Modeling in combination with time Independent Component Analysis for dimension reduction.
Protein motions and dynamics are governed by the underlying free energy. MD simulation are one of the best methods to uncover these underlying hypersurfaces. The biggest challenges are the long timescales needed for obtaining converged (Boltzmann sampled) trajectories and challenge to identify the few functionally important degrees of freedom with the ten-thousands of degrees of freedom. Overcoming this challenges can be real fun.
When studying membrane proteins and their transport function, then only a few process are fast enough to be studied directly with MD simulation without the need of enhanced sampling approaches. Typically such sufficiently fast process can be triggered by ligand binding or release, and can be initiated by placing the ligand into its binding position.
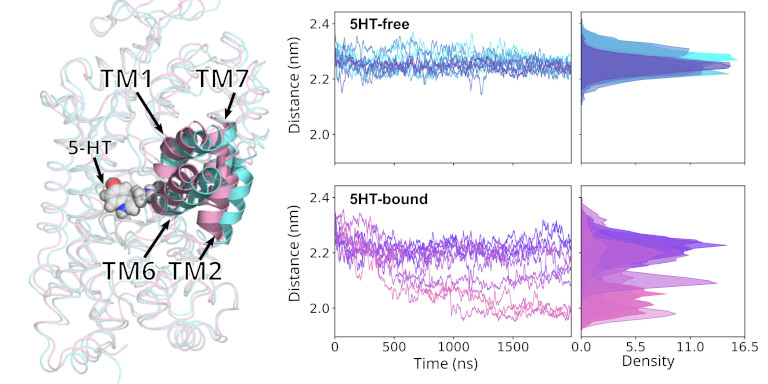
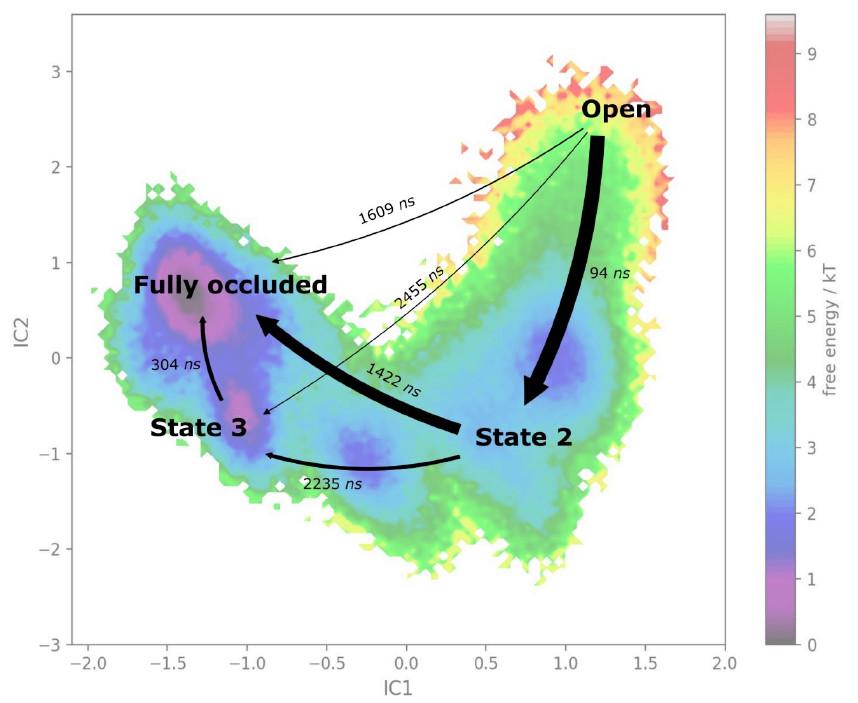
From such trajectories is then possible to reconstruct the underlying free energy using Markov State Modelling approaches, if used in combination with dimension reduction methods. An approach we are using in our lab is time-lagged independent Component Analysis (tICA), which is a supervised learning approach for dimensionality reduction, and we couple this with the Markov State Modelling and Transition State Theory analysis to derive the underlying free energy hypersurface and transition rates.
Many of the biological process are in the milisecond to second timescale and thus too slow for direct simulations and therefore also to uncover the underlying. Also in this cases, the underlying free energy surface of the protein motions can be derived. Trajectories obtained from Milestoning and Swarm simulations, if sufficient in numbers, can also be treated as these unbiased simulations to obtain free energy surfaces.
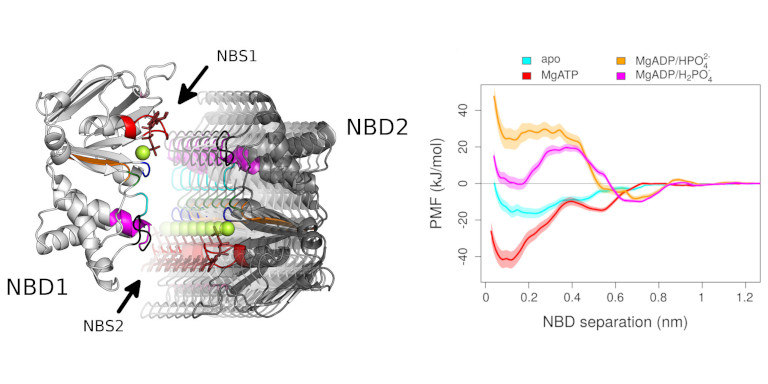
We are using the enhanced sampling methods well-tempered Metadynamics and accelerated weight histogram method (AWH) which allow to describe the conformational change, but also to obtain the underlaying free energy hypersurface. As complementary method, we use of the oldest and very robust perturbation methods, which is the Potential of Mean Force (PMF) approach in combination with the Weighted Historgram Analysis Methods (WHAM) to derive one dimensional free energy profiles.
Experimental approaches
The Stockner lab pairs computational approaches with wet lab experiments. If properly designed, they allow for verification of simulations data, but experimental data also give important insights on transporter function on the macroscopic level beyond the scope of the microscopic lense of single protein MD simulation approaches. Well-designed studies allow to connect the macroscopic data from wet-lab experiments with the microscopic world of computation data, thus leveraging on the resolution and the insights of the complementary approach in a synergistic way to obtain holistic views on protein function.
The most import experimental approaches are pharmacological methods to determine the Michaelis-Menten parameters (Km, Vmax, Bmax, Ki, Kd) of substrates and inhibitors. These methods include the measurement of substrate uptake, inhibition of uptake, inhibitor binding, efflux and the quantification of protein expression. When combined with site directed mutagenesis, these approaches allow for probing the role and importance of individual residues.
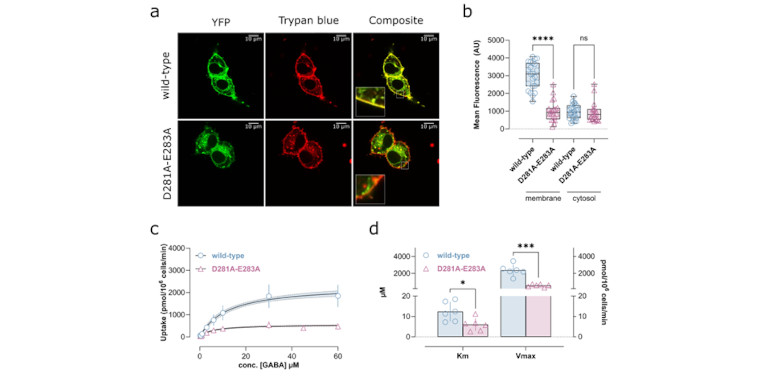
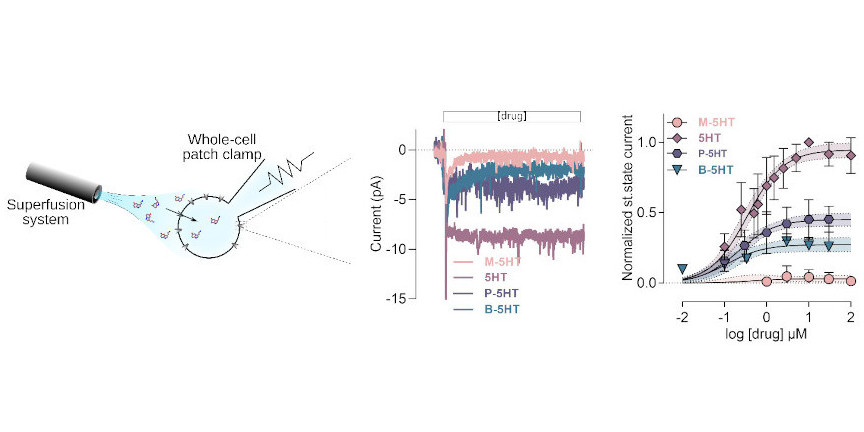
The second experimental field we frequently use is electrophysiology, if substrate translocation is electrogenic, which is true for ion channels and for many of the secondary active transporters we study. Using the patch-clamp approach to measure transmembrane currents, the methods allow for quantifying the amount of charge crossing the cell membrane which directly correlates with transport function.
In addition to experiments carried out in the Stockner Lab, we closely collaborate with a number of experimental groups in house, but also worldwide.

Assoc.-Prof. Dr. Thomas Stockner
ORCID: 0000-0002-7071-8283
Tel.: +43 (0)1 40160-31215
E-Mail: thomas.stockner@meduniwien.ac.at